
Epilepsie: Tierversuche und tierversuchsfreie Forschung
Epilepsie ist eine neurologische Erkrankung, die weltweit etwa 50 Millionen Menschen betrifft. In Deutschland befinden sich etwa 800.000 Menschen aufgrund einer Epilepsie in ärztlicher Behandlung (1). Dabei sind die Therapiemöglichkeiten bisher unbefriedigend und bis zu einem Drittel der Patienten profitiert nicht von den verfügbaren Medikamenten. Um wirksamere Behandlungen zu entwickeln und die Lebensqualität der Patienten zu verbessern, ist weitere Forschung nötig. Doch wie wird sie durchgeführt, und ist sie ethisch vertretbar und wissenschaftlich sinnvoll?
Epilepsie zeichnet sich durch wiederkehrende Anfälle aus, die durch plötzliche, überschießende elektrische Aktivität im Gehirn verursacht werden. Diese Anfälle können sehr unterschiedlich ausfallen – von kurzen Momenten der Abwesenheit bis hin zu schweren Krampfanfällen. Man unterscheidet generalisierte und fokale Anfälle. Bei generalisierten Anfällen ist das gesamte Gehirn betroffen. Sie führen häufig zu Bewusstlosigkeit und Krämpfen im ganzen Körper. Fokale Anfälle entstehen in einem bestimmten Bereich des Gehirns. Je nach Funktion des betroffenen Bereichs kann es zu Zuckungen einzelner Gliedmaßen, Gefühlsstörungen oder Veränderung des Sehens kommen. Auch Halluzinationen und Angstzustände sind möglich (2).
Verfügbare Behandlungsmöglichkeiten
Die Behandlung von Epilepsie umfasst hauptsächlich medikamentöse Therapien, chirurgische Eingriffe und nicht-pharmakologische Ansätze wie die Neurostimulation.
- Medikamentöse Therapie: Antiepileptika sind die erste Wahl bei der Behandlung von Epilepsie. Sie wirken, indem sie die Erregbarkeit von Nervenzellen reduzieren oder die Signalübertragung im Gehirn modulieren. Obwohl viele Patienten gut auf diese Medikamente ansprechen, bleibt etwa ein Drittel der Betroffenen medikamentenresistent (3).
- Chirurgische Eingriffe: Wenn die Anfälle auf eine spezifische Region des Gehirns begrenzt sind, kann bspw. eine operative Entfernung dieser Region die Anfälle deutlich reduzieren oder sogar beseitigen (4).
- Nicht-pharmakologische Ansätze: Dazu gehören die ketogene Diät (5) und Neurostimulationstechnologien wie der Vagusnervstimulator oder die Tiefenhirnstimulation (6).
Trotz dieser vielfältigen Behandlungsmöglichkeiten bleibt die Behandlung von Epilepsie für viele Patienten unbefriedigend. So wirkt die medikamentöse Therapie bei etwa 30 % der Patienten nicht (2). Zudem weisen die Medikamente zum Teil schwere Nebenwirkungen auf. Daher wird weiterhin an der Erkrankung und neuen Behandlungsmethoden geforscht, wofür viele Forscher noch immer Tierversuche einsetzen.
Tierversuche in der Epilepsieforschung
Tierversuche sind seit Jahrzehnten Bestandteil der Epilepsieforschung. Dabei werden vor allem Nagetiere, insbesondere Mäuse und Ratten, eingesetzt, um die Mechanismen der Erkrankung zu verstehen oder neue Therapien zu testen.
Zu den gängigen „Modellen“ gehören genetisch veränderte Tiere oder solche, bei denen Epilepsie-ähnliche Zustände durch chemische Substanzen oder elektrische Reize induziert werden.
Chemische Induktion
Bei der chemischen Induktion werden den Tieren verschiedene Substanzen entweder systemisch oder direkt in das Gehirn injiziert.
Ein Beispiel dafür ist der Wirkstoff Pilocarpin, der eine Übererregung bestimmter Synapsen auslöst und so epileptische Zustände verursacht. Die Substanz wird häufig in die Bauchhöhle injiziert (7) und führt bei Mäusen zu einer Sterblichkeit von 25 bis 100 %, wobei die Tiere häufig an Atemversagen durch Krampfanfälle sterben (8). Um diese hohe Todesrate (Letalität) zu verringern, können die Tiere vor der Pilocarpininjektion Atropin erhalten. Nach der Injektion von Pilocarpin erleiden die Tiere Krampfanfälle, die bei 10 bis 30 % der Tiere zum Tod führen können. Wenn die Tiere länger als 30 Minuten einen generalisierten Anfall erleiden, wird ihnen das Beruhigungsmittel Diazepam verabreicht. Etwa 1 bis 4 Wochen nach der Induktion treten spontan weitere Anfälle auf.
Kaininsäure ist ein weiterer Wirkstoff, der übermäßige neuronale Erregung, insbesondere im Hippocampus, hervorruft. Auch Kaininsäure wird in die Bauchhöhle injiziert, kann jedoch auch direkt ins Gehirn gespritzt werden. Dies führt zu epileptischen Anfällen und kann das Absterben von Nervenzellen verursachen (9).
Auch Tetanustoxin wird verwendet, um bei Tieren epilepsieähnliche Zustände hervorzurufen. Dazu wird es direkt ins Gehirn der Tiere injiziert. Zum Beispiel werden Ratten narkotisiert und ihr Kopf in einem stereotaktischen Rahmen fixiert. Anschließend wird ein Loch in den Schädel gebohrt, durch das eine Nadel in das Gehirn eingeführt wird. Über diese Nadel wird Tetanustoxin in einer kleinen Menge Flüssigkeit in das Gehirn injiziert. 1 bis 3 Tage nach der Injektion erleiden die Tiere wiederkehrende Anfälle (10). Teilweise werden den Tieren zusätzlich Elektroden in das Gehirn implantiert, über die Anfälle beobachtet und vermessen werden können. An den Tieren wird dann untersucht, wie sich verschiedene Wirkstoffe auf die Anfälle auswirken (10).

Mäusen werden Chemikalien gespritzt, die zu Epilepsie-ähnlichen Anfällen führen.
Elektrische Reizung
Bei diesem Ansatz werden Elektroden in das Gehirn der Tiere implantiert, um gezielt elektrische Stimuli zu setzen. Dazu werden Mäuse oder Ratten narkotisiert, ihr Kopf in einen stereotaktischen Rahmen eingespannt und der Schädel aufgebohrt. Durch das Loch im Schädel werden Elektroden ins Gehirn eingeführt und am Schädel befestigt. Nachdem sich die Tiere einige Tage von dem Eingriff „erholt“ haben, wird Strom an die Elektroden angelegt, wodurch die Tiere epileptische Anfälle erleiden.
Beim sogenannten „Kindling-Modell“ (von engl. kindle: etwas anfachen) wird ein Epilepsie-ähnlicher Zustand durch wiederholte, schwache elektrische Stimulation bestimmter Gehirnregionen verursacht. Jeder einzelne Reiz löst zunächst keinen Anfall aus. Mit der Zeit führen diese wiederholten Stimulationen jedoch zu Veränderungen im Gehirn. Das Gehirn wird durch die Reize zunehmend empfindlicher, was zu schwereren und längeren Anfällen führt. Da Forscher glauben, dass dieser Prozess die Entstehung einer Epilepsie nachahmt, wir das Kindling-Modell eingesetzt, um den Mechanismen der Entstehung einer Epilepsie zu untersuchen. Es wird auch verwendet, um die Wirksamkeit von Antiepileptika und anderen Therapien zu testen (11).
Genetische Modelle
Bei diesen „Modellen“ werden Tiere so gezüchtet oder genetisch modifiziert, dass sie spontan Epilepsie-ähnliche Zustände ausbilden.
Häufig werden dabei Ionenkanäle, die eine entscheidende Rolle bei der neuronalen Erregbarkeit spielen, gentechnisch verändert. Bei Scn1a-Knockout (KO) Mäusen wird bspw. der Natriumkanal NaV1.1 gentechnisch ausgeschaltet. Diese Störung betrifft hemmende Neuronen (Interneuronen), was zu einer Übererregbarkeit und einer gesenkten Krampfschwelle führt. Das Ungleichgewicht zwischen erregenden und hemmenden Signalen im Gehirn führt zu schweren Anfällen (11).
Auch werden Mäuse gentechnisch so verändert, dass sie bestimmte Mutationen aufweisen, die beim Menschen mit Epilepsie in Verbindung stehen. Beispielsweise führen Mutationen im PTEN-Gen, die beim Menschen mit tuberöser Sklerose assoziiert sind, zu abnormalem Zellwachstum und verursachen unter anderem Fehlbildungen im Gehirn. Dadurch werden neuronale Schaltkreise gestört und Epilepsie-ähnliche Zustände hervorgerufen (11).
Neben den hier exemplarisch vorgestellten Beispielen gibt es eine Vielzahl an weiteren gentechnisch veränderten „Tiermodellen“. Dabei handelt es sich überwiegend um gentechnisch veränderte Mäuse, da die Techniken zur genetischen Manipulation bei Mäusen weit fortgeschritten und gut verfügbar sind. Aber auch gentechnisch veränderte Zebrafische werden in der Epilepsieforschung genutzt. (11).
Weitere Modelle
Bei Gerbils können Epilepsie-ähnliche Zustände ausgelöst werden, indem die geräuschempfindlichen Tiere intensiven Geräuschen ausgesetzt werden (11). Auch Paviane werden in der Epilepsieforschung verwendet (12). Bei den Tieren werden Epilepsie-ähnliche Anfälle durch Stimulation mit Stroboskoplicht (Flackerlicht) ausgelöst. Während der Stimulation können generalisierte Anfälle auftreten, die 20 bis 30 Sekunden andauern (11).
Bei allen sogenannten Epilepsie-Modellen ist das Leid der Tiere immens. Neben den akuten Anfällen leiden viele unter chronischen neurologischen und körperlichen Problemen, die durch die induzierten Zustände entstehen.
Das Scheitern der tierversuchsbasierten Epilepsieforschung
Die Erfolgsquote bei der tierversuchsbasierten Medikamentenentwicklung, also die Wahrscheinlichkeit, dass ein Wirkstoff, der in Tierversuchen als wirksam und sicher getestet wurde, auch in klinischen Studien am Menschen wirkt und eine Zulassung erhält, liegt im Durchschnitt über alle Indikationen bei unter 7 % (13).
Besonders schlecht schneiden dabei mit einer Erfolgsrate von nur 5,3 % Medikamentenentwicklungen im Bereich der Neurologie – zu dem auch Epilepsie gehört – ab (13). Das bedeutet, dass beinahe 95 % der Substanzen zur Behandlung neurologischer Erkrankungen trotz vermeintlich positiver Ergebnisse in Tierversuchen in den klinischen Studien am Menschen scheitern. Die meisten Wirkstoffe scheitern dabei in Phase II der klinischen Studien (14), also zu dem Zeitpunkt, an dem die Wirksamkeit erstmals an erkrankten Menschen getestet wird. Dies belegt, dass die präklinische Forschung, die maßgeblich auf Tierversuchen basiert, aufgrund mangelnder Übertragbarkeit auf den Menschen versagt.
Im Folgenden werden die Gründe für die mangelnde Übertragbarkeit von Tierversuchen auf den Menschen im Bereich der Epilepsieforschung kurz zusammengefasst.
Unterschiede zwischen den Spezies
Die Physiologie und das Gehirn von Tieren unterscheiden sich erheblich von denen des Menschen. Viele Ergebnisse aus Tierversuchen lassen sich daher nicht auf den Menschen übertragen. So zeigen Medikamente, die bei Tieren wirksam sind, häufig keine Wirkung oder unerwartete Nebenwirkungen beim Menschen.
Das Gehirn von Nagetieren, die am häufigsten in der Epilepsie-Forschung verwendet werden, unterscheidet sich in Größe, Struktur und neuronaler Organisation deutlich vom menschlichen Gehirn. So ist der Hippocampus von Mäusen im Verhältnis zur Gesamthirnmasse größer, während das menschliche Gehirn eine ausgeprägtere kortikale Komplexität aufweist. Beim Menschen spielt der Neokortex eine zentrale Rolle bei vielen Epilepsien, während bei Nagetieren Epilepsie häufig im Hippocampus „modelliert“ (also künstlich verursacht) wird. Somit entspricht das Umfeld, in dem Epilepsie in Tieren untersucht werden soll, nicht der Situation im Gehirn erkrankter Menschen.
In genetischen „Tiermodellen“ werden einzelne Gene manipuliert, um genetische Epilepsieformen zu simulieren. Diese Modelle ahmen somit bestenfalls monogene Epilepsien nach, die auf der Veränderung eines einzelnen Gens beruhen. Beim Menschen sind solche monogenen Epilepsien jedoch selten (15). Stattdessen sind Epilepsien beim Menschen oft polygenetisch oder multifaktoriell, was durch Tierversuche nicht dargestellt werden kann.
Künstliche Krankheitsmodelle
Die bei Tieren induzierten Epilepsie-ähnlichen Zustände unterscheiden sich grundlegend von der menschlichen Erkrankung. Beispielsweise fehlt es oft an der Komplexität und der Vielfalt, die menschliche Epilepsie auszeichnet.
Epilepsie wird in Tiermodellen künstlich induziert, z. B. durch chemische (Pilocarpin, Kaininsäure) oder elektrische Reizung oder durch genetische Manipulation. Diese Methoden simulieren bestimmte Aspekte der Epilepsie (z. B. fokale oder generalisierte Anfälle), spiegeln jedoch nicht die auf vielen Ursachen beruhende menschliche Epilepsie wider.
Epileptische Anfälle bei Tieren zeigen oft ein gleichförmiges Muster, das durch die induzierenden Methoden vorgegeben wird (z. B. durch chemische Induktion). Diese Anfälle können in ihrer Dauer und Intensität von menschlichen Anfällen abweichen. Menschen erleben dagegen ein breiteres Spektrum an Anfallsarten, die sich in Länge und Intensität deutlich unterscheiden können. Die Anfallsdynamik beim Menschen wird zudem durch emotionale Faktoren und äußere Reize beeinflusst, was in Tierversuchen kaum adäquat berücksichtigt werden kann.
Chemische und elektrische Induktionen führen oft zu einem akuten Anfallsstatus und anschließend zu einer Phase der chronischen Epilepsie. Dabei treten nach Wochen oder Monaten spontane Anfälle auf, jedoch in vorhersehbaren Mustern. Beim Menschen können Anfälle dagegen spontan auftreten, oft ohne klare Auslöser. Der chronische Verlauf und die Unvorhersehbarkeit der Anfälle sowie das breite Spektrum der Symptome machen die menschliche Epilepsie somit komplexer.
Neben den wissenschaftlichen Gründen, die das Scheitern der tierversuchsbasierten Epilepsieforschung erklären, gibt es weitere Argumente gegen Tierversuche. In der Epilepsieforschung werden Tieren bewusst erhebliche Leiden zugefügt. Zudem sind Tierversuche kostenintensiv und zeitaufwendig, während ihre Erfolgsquote bei der Übertragung der Ergebnisse auf den Menschen enttäuschend niedrig bleibt. Dies wirft die grundlegende Frage auf, ob Tierversuche den Fortschritt in der Epilepsieforschung nicht nur nicht fördern, sondern möglicherweise sogar behindern und so mitverantwortlich für das Ausbleiben bedeutender Erfolge sind.
Daher gibt es zunehmend Bestrebungen, tierversuchsfreie Methoden für die Epilepsieforschung zu entwickeln.
Tierversuchsfreie Forschungsmethoden
Moderne Zellkulturen, insbesondere dreidimensionale Gehirnorganoide, erlauben es, menschliches Gehirngewebe in vitro zu untersuchen. Diese Modelle können auch genetische und biochemische Eigenschaften von Epilepsiepatienten erfassen.
Zellkulturen
Im einfachsten Fall werden Zellkulturen von Neuronen verwendet. Bisher stammten diese Zellen oft aus getöteten Tieren, da menschliche Neuronen aus ethischen Gründen nur schwer erhältlich sind. Dank humaner induzierter pluripotenter Stammzellen (iPSCs) ist es heute jedoch möglich, menschliche Neuronen herzustellen. Dafür können zum Beispiel Hautzellen von Menschen verwendet und in iPSCs „zurückprogrammiert“ werden. Aus diesen iPSCs lassen sich durch gezielte Differenzierung verschiedene Zelltypen erzeugen, darunter auch Neuronen.
Der Vorteil dieser Methode liegt darin, dass direkt an menschlichen Zellen geforscht werden kann, die außerdem von Patienten gewonnen werden können. Das macht aus iPSCs abgeleitete Neuronen besonders geeignet für die Untersuchung genetisch bedingter Epilepsieformen und bietet die Möglichkeit, Therapien individuell an Patienten anzupassen (16).
Allerdings fehlt einfachen Zellkultursystemen die strukturelle Komplexität des Gehirns, in dem verschiedene Zelltypen miteinander agieren. Daher werden zunehmend dreidimensionale Zellkulturen wie beispielsweise sogenannte Organoide in der Epilepsieforschung eingesetzt.
Organoide
Hirnorganoide sind dreidimensionale Strukturen, die aus iPSCs gewonnen werden können. Sie können den Aufbau und die zelluläre Vielfalt des menschlichen Gehirns nachahmen. Dazu werden iPSCs in neuronale Vorläuferzellen umgewandelt, die sich dann von selbst zu komplexen Strukturen organisieren. Diese Strukturen ähneln verschiedenen Gehirnregionen und der Prozess ihrer Bildung ahmt die Entwicklung des menschlichen Gehirns im Embryo nach (17).
Diese Hirnorganoide können neuronale Netzwerke bilden und weisen funktionelle Eigenschaften von echtem Gehirngewebe auf. Hirnorganoide sind besonders hilfreich, um die zellulären und molekularen Mechanismen hinter Epilepsie zu erforschen. Sie ermöglichen es Wissenschaftlern, zu untersuchen, wie bestimmte genetische Mutationen die Entwicklung, Vernetzung und Erregbarkeit von Neuronen beeinflussen. Außerdem können Organoide genutzt werden, um die Wirkung potenzieller Medikamente auf die Anfallsaktivität zu testen (17).
An Hirnorganoiden lassen sich auch elektrophysiologische Messungen durchführen (17). Wu et al. (2022) haben Hirnorganoide aus Stammzellen von zwei Patienten mit einer neurologischen Entwicklungsstörung, die durch Mutationen im CDKL5-Gen verursacht wird (CDD), entwickelt. CDD-Patienten zeigen Entwicklungsverzögerungen, früh einsetzende Krampfanfälle und autistisches Verhalten, was auf eine Übererregbarkeit der Nervenzellen zurückzuführen ist. Mit speziellen elektrophysiologischen Techniken analysierten Wu et al., wie Nervenzellen in diesen Hirnorganoiden Signale weiterleiten und elektrische Impulse erzeugen, und verglichen sie mit gesunden Kontrollproben. Die Organoide zeigten eine ähnliche Übererregbarkeit wie bei CDD-Patienten. Es konnte gezeigt werden, dass die Störung der Ionenkanäle eine zentrale Rolle bei der Entstehung der Krankheit spielt (18).
Neben der Untersuchung von Krankheitsmechanismen lassen sich Hirnorganoide auch in der Medikamentenentwicklung einsetzen. So nutzten Yokoi et al. (2021) eine spezielle Methode, bei der sie Hirnorganoide mit einem Mehrfachelektrodenarray kombinierten. Mit diesem Ansatz untersuchten sie die Reaktion der Organoide auf krampfauslösende Substanzen und auf verschiedene Medikamente mit unterschiedlichen Wirkmechanismen. Die Methode erwies sich als nützlich, um sowohl das unerwünschte Krampfpotenzial von Medikamenten zu ermitteln als auch die Aktivitätsänderungen der Organoide nach Verabreichung von Antiepileptika zu bewerten (19).
Hirnorganoide stellen somit ein modernes und am Menschen ausgerichtetes Werkzeug in der Epilepsieforschung dar, das tiefe Einblicke in menschenspezifische Krankheitsmechanismen ermöglicht, die in sogenannten Tiermodellen nicht erfasst werden können.
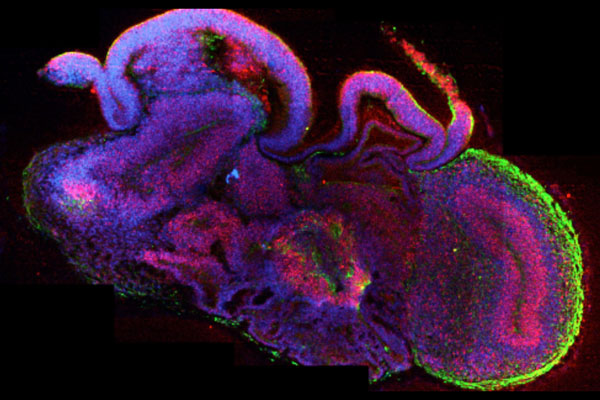
Hirnorganoide bilden die Strukturen des menschlichen Gehirns nach und weisen funktionelle Eigenschaften auf.
©Madeline Lancaster (Lancaster, M.A. Nature 2013; 501: 373-379)
Organ-on-a-Chip Systeme
Ein mikrophysiologisches System (MPS) ist ein künstlich hergestelltes Modell aus zwei- oder dreidimensionalen Zellstrukturen, die als Organ-on-a-Chip bezeichnet werden. Diese „Chips“ enthalten lebende Zellen, die unter kontrollierten Bedingungen und einem kontinuierlichen Flüssigkeitsfluss kultiviert werden. Dadurch lassen sich die physiologischen und krankheitsbedingten Funktionen menschlicher Organe nachbilden.
Beispielsweise entwickelten Pelkonen et al. (2020) ein Epilepsie-on-a-Chip System. Auf dem Chip werden drei separate neuronale Netzwerke miteinander über feine mikrofluidische Kanäle verbunden. Die Aktivität der Netzwerke kann mit speziellen Mikroelektroden überwacht werden. Die Netzwerke zeigten spontane Aktivitätsmuster, die sowohl lokal als auch zwischen den miteinander verbundenen Netzwerken synchron waren, ähnlich den Gehirnfunktionen. Bei Zugabe der Anfall-auslösenden Substanz Kaininsäure verstärkten sich die Aktivitäten nur in den behandelten Netzwerken. Das Antiepileptikum Phenytoin konnte die Aktivität der übererregten Netzwerke erfolgreich reduzieren (20). Das entwickelte System ermöglicht erstmals die Modellierung fokaler Anfälle in menschlichen Nervenzellnetzwerken und bietet eine neue Plattform zur Erforschung von Epilepsie und potenziellen Therapien.
Diese innovative Technologie könnte die Medikamentenentwicklung erheblich verbessern und effizienter gestalten und so Tierversuche in der präklinischen Forschung ersetzen (21).
Computermodelle und KI
Die Analyse großer Datenmengen, die durch die Untersuchung komplexer Gewebe und Organe entsteht, erfordert moderne Technologien wie künstliche Intelligenz (KI) und maschinelles Lernen (ML). KI-gestützte Algorithmen können kontinuierliche Daten, z. B. aus EEG-Aufzeichnungen, analysieren, um das Risiko von Anfällen vorherzusagen. Dies ermöglicht es Patienten und Ärzten, vorbeugende Maßnahmen zu ergreifen. Auch ermöglichen KI/ML-Modelle anhand von Patientendaten wie genetischen Informationen, Therapiehistorie und klinischen Merkmalen vorherzusagen, wie ein Patient auf bestimmte Antiepileptika reagieren wird (21).
Schließlich lässt sich KI auch zur Analyse großer Datenmengen, wie sie bspw. bei der Analyse von Organ-on-a-Chip Versuchen entstehen, einsetzen. So kann die Kombination moderner In-vitro-Methoden mit KI/ML zur Entwicklung neuer Therapien beitragen (21).
Während sich die Ergebnisse von Tierversuchen aufgrund der Speziesunterschiede und der Unterschiede zwischen künstlich hervorgerufenen epilepsieähnlichen Zuständen und der tatsächlichen Epilepsie menschlicher Patienten nicht auf den Menschen übertragen lassen, ermöglichen tierversuchsfreie Methoden die Untersuchung direkt am Menschen oder an menschlichem Material. Zudem ermöglichen sie nicht nur für Menschen relevante Ergebnisse, sondern sogar eine auf einzelne Patienten abgestimmte Therapie.
Ursachenforschung
Die genauen Entstehungswege für Epilepsie sind bisher noch nicht vollständig bekannt, was vor allem auch an den in der Forschung eingesetzten ungeeigneten Modellsystemen liegt (17). Mögliche Ursachen für Epilepsie sind genetische Faktoren, Hirnverletzungen, Gehirntumore, Schlaganfälle, Infektionen und mangelnde Sauerstoffversorgung bei Neugeborenen. Tierversuchsfreie Methoden wie genomweite Assoziationsstudien und die Analyse von Patientendatenbanken haben zur Identifizierung genetischer Risikofaktoren beigetragen.
Die Prävention von Epilepsie umfasst entsprechend der Krankheitsursachen vor allem Ansätze zur verbesserten pränatalen Versorgung, Vermeidung von Kopfverletzungen und Kontrolle von Infektionen.
Jenseits dieser primären Präventionsmaßnahmen, die eine Erkrankung zwar möglicherweise verhindern können, jedoch kaum von Individuen beeinflussbar sind, können bereits an Epilepsie erkrankte Personen die Häufigkeit von Anfällen durch Anpassung ihres Lebensstils senken. In diesem Zusammenhang listet die Epilepsy Foundation eine Reihe von Anfalls-Triggern auf, die sich vermeiden lassen. So kann Alkoholkonsum zu Anfällen führen. Auch Schlafmangel und Stress können Anfälle auslösen. Betroffene Patienten können so durch eine gesunde Lebensweise die Anzahl der Anfälle reduzieren (22).
Fazit
Die Epilepsieforschung steht an einem Wendepunkt. Während in der Vergangenheit Epilepsie-Forschung hauptsächlich tierexperimentell betrieben wurde, wird zunehmend klar, dass Tierversuche wissenschaftlich nicht zielführend und ethisch problematisch sind. Moderne, tierversuchsfreie Methoden sind nicht nur effizienter, sondern auch besser geeignet, die Komplexität der menschlichen Epilepsie abzubilden. Die Fortschritte in der personalisierten Medizin, wie die Nutzung von iPSCs, ermöglichen es, patientenspezifische Modelle zu erstellen und individuelle Therapien zu entwickeln.
Ein Paradigmenwechsel hin zu tierversuchsfreien Ansätzen könnte nicht nur Tierleid verhindern, sondern auch dazu beitragen, bessere Behandlungs- und Präventionsmöglichkeiten für Millionen von Patienten weltweit zu entwickeln.
20.01.2025
Dr. rer. nat. Johanna Walter
Weitere Infos
Quellen
- Deutsche Epilepsievereinigung: Aktuelle Daten zur Epilepsie und zum Behandlungsstand
- Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG): Epilepsie
- Janmohamed M. et al. Pharmacoresistance – epidemiology, mechanisms, and impact on epilepsy treatment. Neuropharmacology 2020; 168:107790
- Sabzvari T. et al. A comprehensive review of recent trends in surgical approaches for epilepsy management. Cureus 2024; doi: 10.7759/cureus.71715
- Barañano K.W. et al. The ketogenic diet: Uses in epilepsy and other neurologic illnesses. Current Treatment Options in Neurology 2008; 10(6):410–419
- Simpson H.D. et al. Practical considerations in epilepsy neurostimulation. Epilepsia 2022; 63(10):2445–2460
- Curia G. et al. The pilocarpine model of temporal lobe epilepsy. Journal of Neuroscience Methods 2008; 172(2):143–157
- Buckmaster P.S. et al. Factors affecting outcomes of pilocarpine treatment in a mouse model of temporal lobe epilepsy. Epilepsy Research 2012; 102(3):153–159
- Rusina E. et al. The kainic acid models of temporal lobe epilepsy. eneuro 2021; 8(2):ENEURO.0337-20.2021
- Doheny H.C. et al. A comparison of the efficacy of carbamazepine and the novel anti‐epileptic drug levetiracetam in the tetanus toxin model of focal complex partial epilepsy. British Journal of Pharmacology 2002; 135(6):1425–1434
- Rubio C. et al. Classification of current experimental models of epilepsy. Brain Sciences 2024; 14(10):1024
- Szabó C.Á. et al. The baboon in epilepsy research: Revelations and challenges. Epilepsy & Behavior 2021; 121:108012
- Why are clinical development success rates falling? Biomedtracker, April 2024
- Clinical development success rates and contributing factors 2011–2020. Biomedtracker, Februar 2021
- Boßelmann C. et al. Genetische Diagnostik der Epilepsien: Empfehlung der Kommission Epilepsie und Genetik der Deutschen Gesellschaft für Epileptologie (DGfE). Clinical Epileptology 2023; 36(3):224–237
- Weng O.Y. et al. Modeling epilepsy using human induced pluripotent stem cells-derived neuronal cultures carrying mutations in ion channels and the mechanistic target of rapamycin pathway. Frontiers in Molecular Neuroscience 2022; 15:810081
- Brown R. et al. Progress and potential of brain organoids in epilepsy research. Stem Cell Research & Therapy 2024; 15(1):361
- Wu W. et al. Neuronal hyperexcitability and ion channel dysfunction in CDKL5-deficiency patient iPSC-derived cortical organoids. Neurobiology of Disease 2022; 174:105882
- Yokoi R. et al. Analysis of signal components < 500 Hz in brain organoids coupled to microelectrode arrays: A reliable test-bed for preclinical seizure liability assessment of drugs and screening of antiepileptic drugs. Biochemistry and Biophysics Reports 2021; 28:101148
- Pelkonen A. et al. A modular brain-on-a-chip for modelling epileptic seizures with functionally connected human neuronal networks. Biosensors and Bioelectronics 2020; 168:112553
- Shariff S. et al. Tailoring epilepsy treatment: personalized micro physiological systems illuminate individual drug responses. Annals of Medicine & Surgery 2024; doi: 10.1097/MS9.0000000000002078
- Epilepsy foundation: Seizure Triggers